Batch Manufacturing Record Guidelines in the Pharmaceutical Industry
Follow these Batch Manufacturing Record Guidelines to avoid 483s digitize documentation ensure data integrity and meet evolving GMP expectations in 2025

Why BMRs Matter More Than Ever in 2025
In today’s pharmaceutical world Batch Manufacturing Records (BMRs) are more than just a regulatory requirement—they’re a key part of modern compliant manufacturing. As we move into 2025 the role of BMRs has grown exponentially. No longer confined to paper binders or static forms BMRs now sit at the intersection of digital transformation, real-time compliance and data driven quality management. Tracking quality control tests is key to having complete visibility of the manufacturing process so you can make informed decisions based on fact.


Global regulators (FDA, EMA, WHO) are raising the bar on documentation practices and remote audits, AI enhanced quality systems and connected shop floors have made the traditional reactive approach to batch records obsolete. For manufacturers who want to stay competitive and inspection ready BMRs must now evolve into dynamic tools that document the entire batch process and drive efficiency and traceability across the product lifecycle.
Explore FDA-aligned batch record frameworks designed to meet GMP expectations without complexity.
Strategic Role of Batch Manufacturing Records in GMP Compliance
At their core Batch Manufacturing Records (BMRs) are not just a documentation requirement—they are a compliance mechanism to safeguard product quality, ensure regulatory compliance, protect patient safety and uphold data integrity through rigorous quality control.
Anchored in Global GMP Principles:
Whether you’re following FDA 21 CFR Part 211, EU GMP Annex 11 or WHO GMP guidelines BMRs are key to demonstrating:
- Every batch was manufactured according to the approved procedures (as outlined in the MFR)
- All critical process parameters (CPPs) and in-process controls (IPCs) were followed
- Any deviation was identified, recorded and resolved with full traceability
- Quality control procedures were implemented to comply with GMP principles and regulatory specifications
The Compliance Edge:
A strategic BMR framework:
- Prevents undocumented process drift so you comply and reduce batch failures
- Ensures data integrity through validated entries, signatures and real-time capture
- Creates a single source of truth for internal QA teams and external regulators. When done correctly BMRs are your first line of defence during regulatory inspections—and your strongest proof of operational control.
How Master Formula Records (MFR) and BMRs Interact
The Master Formula Record (MFR) defines how a product should be made. The Batch Manufacturing Record (BMR) documents how it was actually made—for a specific batch. The batch number is the unique identifier for each production batch, quality control and regulatory compliance.
In short the MFR is the “ideal” process—what should be done, in what order and under what conditions. It includes formulas, processing instructions and in-process controls. The master batch record is the foundation document for the ideal production process containing detailed instructions on raw materials, equipment and conditions for manufacturing. The BMR captures what actually happened during a specific batch. It includes data entries, operator signatures, deviations, environmental readings and yield calculations.
Here’s the seamless flow:
MFR -> BMR -> Review & Release
- The MFR sets the standard: predefined ingredients, steps, equipment and controls.
- The BMR captures execution in real time: operator inputs, deviations, yield and QC results.
- The QA/QC review ensures the batch matches the approved protocol before it’s released.
This link between MFR and BMR is a closed loop of traceability so every action is verifiable, auditable and compliant—enabling strategic manufacturing control.
Review & Approval: Closing the Loop with Quality
Traditionally, BMRs were reviewed by the quality assurance (QA) team days or even weeks after batch completion. This often introduced delays in product release and increased the risk of missed discrepancies. Today, that model is being disrupted. Quality control testing is now an integral part of ensuring compliance with health and safety regulations.
In 2025, batch record review isn’t just about compliance—it’s about accelerating time-to-release without compromising data integrity. Modern quality teams are turning to automation, smart validation, and real-time collaboration to make the review process faster, more accurate, and inspection-ready at all times. Detailed audit trails, including timestamps and user identifications, are essential for maintaining regulatory standards and tracking modifications throughout the manufacturing process.
Learn how automated batch release processes reduce QA review time and ensure compliance from draft to release.
Real-World Review Workflows Today:
- Parallel QA review while production is ongoing
- Automated deviation flags and exception based review
- Pre-validation of critical entries based on SOP linked rules
- Quality control checks to ensure the product meets established quality standards
- Tracking information about personnel involved in each production step
Powered by Digital Tools:
- AI assisted review identifies anomalies, missing fields or out of spec entries
- Smart validation checklists ensure all critical steps are completed and verified
- Digital sign-offs streamline multi level approval hierarchies with role based access
- Electronic batch records streamline the review process, reduce human error and improve accuracy
- Master packaging records outline packaging instructions after the manufacturing process, ensuring compliance and efficiency
This modern workflow not only speeds up batch release—it builds a culture of continuous quality where compliance is embedded in the process not just a post-facto check.
Close the quality loop with digital clarity. See Leucine’s real-time review system in action
What Happens When BMRs Go Wrong
When Batch Manufacturing Records (BMRs) fail the consequences can be severe—FDA warning letters, product recalls, supply chain delays and worst of all patient safety risks. These failures often share one common root: poor documentation practices. Ensuring detailed records is critical for compliance with safety and performance standards as they provide a comprehensive overview of the entire production process.
Having accurate and accessible records is essential for regulatory compliance as it supports quality control during production and ensures adherence to regulatory requirements thereby enhancing product safety and effectiveness.


Real-World Case Insights:
- FDA 483 issued in 2024 for a CDMO for incomplete BMR entries, missing signatures and unlogged deviations—batch rejected worth $2 million.* Another company faced regulatory scrutiny when backdated entries were found during an inspection, undermining their entire QA system.
- A firm improved product quality by managing quality testing data and producing high quality products and customer satisfaction.
- Documenting in-process testing results in the Batch Manufacturing Record helped another company meet safety and quality standards throughout the manufacturing process.
Common BMR Failures:
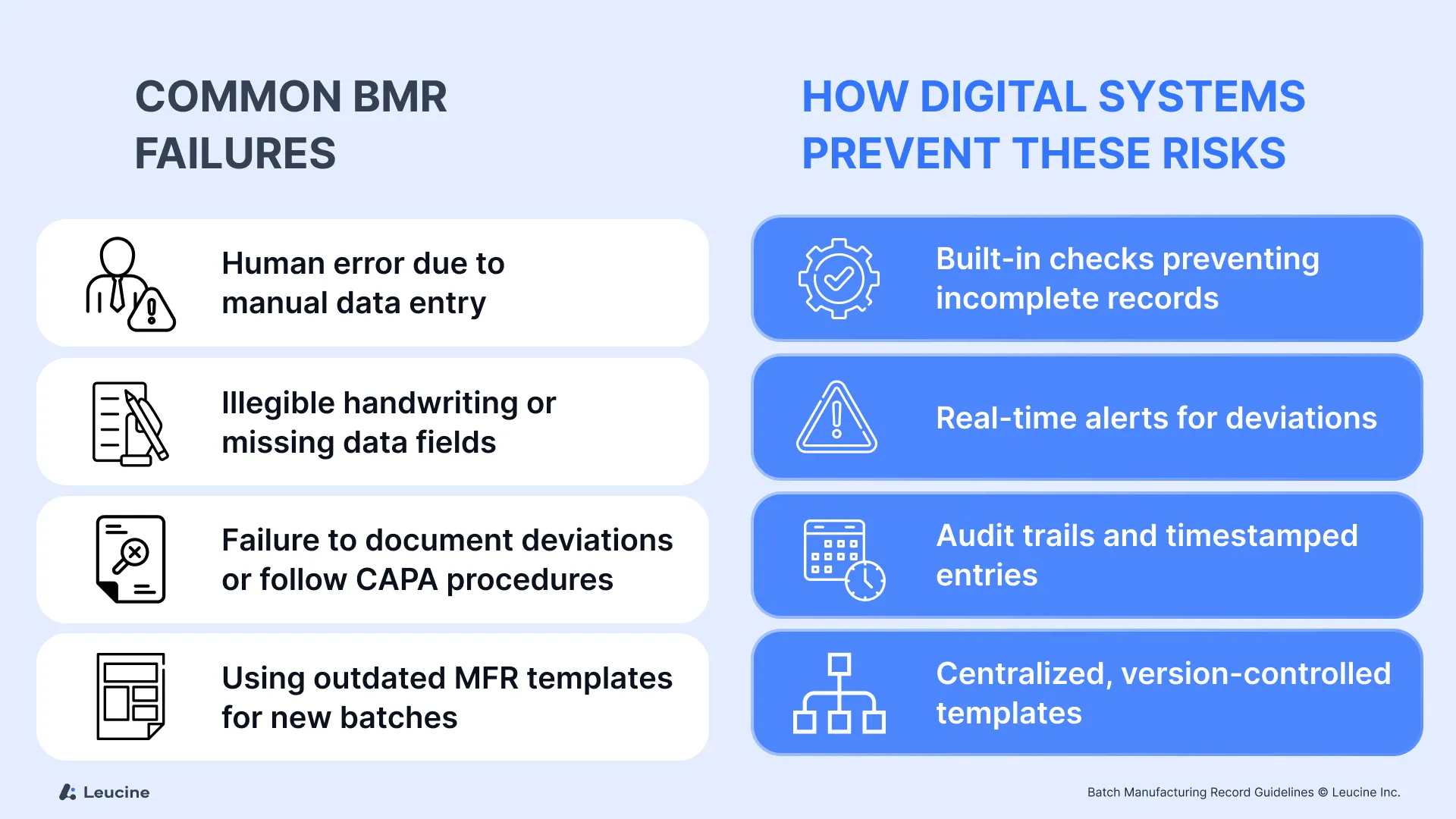
- Human error due to manual data entry
- Illegible handwriting or missing data fields
- Failure to document deviations or follow CAPA procedures
- Using outdated MFR templates for new batches
- Inadequate batch production records for documenting the actual production of each batch
- Incomplete manufacturing instructions for the step-by-step process of making a product
How Digital Systems Prevent These Risks:
- Built-in checks to prevent submission of incomplete records
- Real-time alerts for deviations or out of spec conditions
- Audit trails and timestamped entries that cannot be edited retroactively
- Centralized, version controlled templates to ensure every batch starts with the correct master formula record
- Electronic batch records to reduce human error and improve accuracy
- Audit trail to maintain regulatory standards and track changes
In short paper BMRs create blind spots. Digital systems create transparency.
Protect your operations from hidden risks—switch to a validated eBMR system. Audit your current process with Leucine
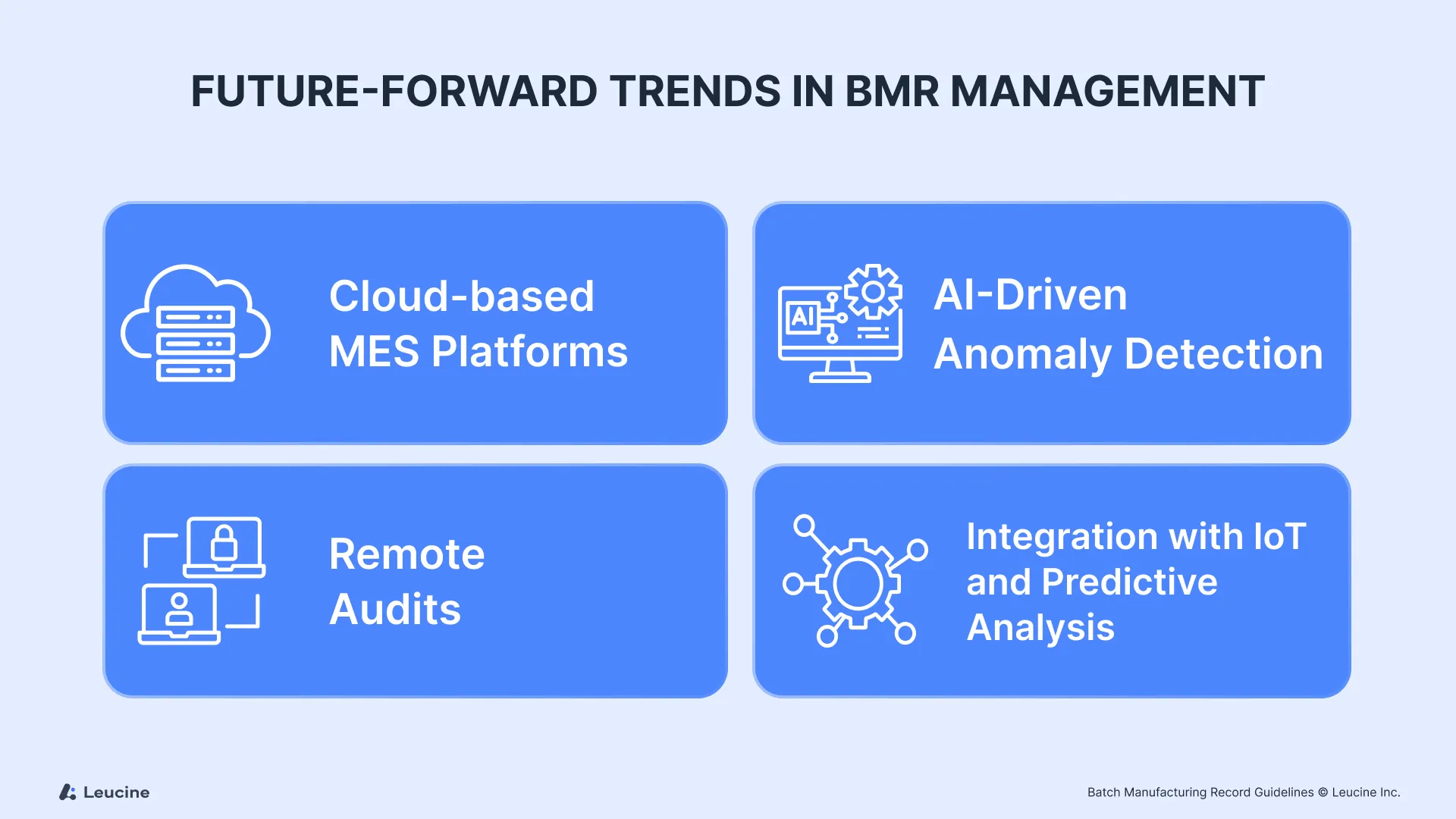
Future-Forward Trends in BMR Management
Looking ahead the next generation of BMRs will go beyond digitization. They will be smart, integrated and predictive, leveraging technologies like AI, machine learning and IoT. Sensors on the shop floor will feed real-time data into BMRs, enabling automated tracking of temperature, humidity, equipment performance and material usage. Integration of process manufacturing equipment with specialized BMR systems will enhance automated data collection from devices like scales and pressure sensors.
As pharmaceutical manufacturing enters a new digital decade Batch Manufacturing Records (BMRs) are at the forefront of intelligent operations and predictive quality management. The future is paperless, connected and proactive, ensuring products meet specific safety and performance criteria by adhering to strict quality standards.
Cloud-Based MES Platforms
- Secure
- Scalable
- Accessible from anywhere
- Enable multi-site standardization and global collaboration
- Real-time synchronization between MFRs, BMRs, QMS and ERP systems
- Increase manufacturing flexibility with software solutions
- Meet regulatory requirements to support compliance
AI-Driven Anomaly Detection
- Uses historical batch data to detect outliers before they become deviations
- Identifies trends in operator behavior, environmental variables or process variability
- Assists in root cause analysis and preventive action recommendations
- Tracks product quality through accurate data collection and automated quality control
- Verifies health and safety procedures are followed through quality control testing to meet FDA requirements
Remote Audits & Regulatory Expectations
- Cloud hosted BMRs for 24/7 audit access for global regulators
- Virtual inspections and remote QA reviews will be standard post 2024
- Compliance with health and safety regulations by following regulatory standards
- Detailed records to meet regulatory requirements
Integration with IoT and Predictive Analytics
- IoT sensors feed live data into BMRs—temperature, humidity, RPM etc.
- Predictive analytics flags potential failures before they occur for proactive batch control
- Quality control ensures products meet regulatory specs and safety standards during manufacturing
- In-process testing documents the quality and compliance of the manufacturing process to ensure each product meets safety and quality standards
These future facing capabilities turn BMRs from static documents into dynamic compliance ecosystems where every entry informs action and every trend informs strategy.

Conclusion: Your Batch Records Are Only as Good as Your Systems
In an age of intelligent manufacturing the BMR is no longer just a compliance checkbox—it’s a strategic tool for quality, risk and operational excellence. Companies that continue to use paper or outdated systems put themselves at a disadvantage not only in compliance but also in speed, scalability and competitiveness. The future of pharma manufacturing will be digital by default and your batch records must evolve to reflect that reality. Batch Manufacturing Records (BMRs) are no longer just documentation—they are strategic tools for quality, regulatory compliance and operational agility. Whether it’s aligning with global GMP frameworks, real-time oversight or AI enabled manufacturing your BMR strategy reflects the maturity of your systems.
Paper records can’t keep up with regulatory expectations, process complexity or digital innovation. But with the right platform you can:
- Compliance by design
- Faster time to release
- Digital first quality culture
In modern pharma your detailed records are your proof of performance—and your platform is the engine that powers them.
Let’s pioneer your digital transformation journey with precision and purpose.
Book your Leucine demo today